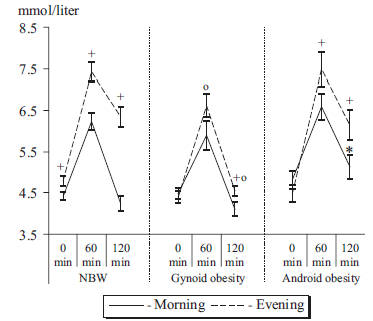
2) gynoid obesity (stores more fat in hips & butt), defined by WC/HC < 0.85
3) android obesity (stores more fat in belly, which is rare in women), defined by WC/HC > 0.85
First, we get confirmation that insulin sensitivity (IS) is better in morning than evening. But then we get these interesting glucose tolerance curves:
Fat stored in your hips & butt is thought to be healthier than that stored in your belly region. This is confirmed here. Gynoid obesity, while exhibiting an attenuated AM/PM difference, was able to restore euglycemia by the end of the experiment at both time points. Ie, gynoid obesity selectively improved IS in the evening.
Android obesity, which is more nefarious than gynoid (also confirmed here), had a similar though not as robust effect in the evening but deteriorated IS in the morning.
One potential interpretation: it’s better to have a little extra fat stored in your hips and butt than to be lean or have belly fat. However, I have a qualm with that interpretation. Healthy people show a robust circadian difference in glucose tolerance. Just as insulin resistance (IR) is an accepted physiological phenomenon observed in some ketogenic dieters, I view this circadian difference, also, as physiological.
I know I know, it was mice fed standard rodent chow, but also included models relevant to human biology like reduced insulin and caloric restriction, which may reflect certain aspects of ketogenic diets and intermittent fasting… and some of the results actually do reflect what happens to humans. Some.
The Common Response: 1) IR -> 2) hyperinsulinemia -> 3) more insulin = more fat mass.
However, this is flawed.
Easiest rebuttal (somewhat of a strawman, but whatevs): Barbara Corkey and her group has done a lot of work showing that insulin hypersecretion (caused by dietary additives, preservatives, weird chemicals, etc.) may actually precede & causes IR… not enough insulin hypersecretion to induce hypoglycemia, just enough to induce IR.
So that basically breaks the 1st step in the Common Response, but doesn’t really disprove the possibility that IR still causes obesity (or can cause obesity).
side note: please consider the modern views of Taubes, Lustig, Gardner, Attia, and others on Carbs™. They’re less “Carbs-cause-obesity, keto-for-all, etc.,” and more thinking it might not be Carbs™ per se, but rather processed and refined foods. And #context… And I tend to agree at the moment (nuances and caveats are subject to change, as more evidence accumulates).
disclaimer: I haven’t seen the full text of Hall’s recent study, but that’s not really relevant to what I want to discuss. In other words, I don’t think the full text will provide any additional details on this particular point.
Tl;dr: this study was not designed to prove or disprove metabolic advantage or the insulin-obesity hypothesis.
It’s in the study design: four weeks of low fat followed by four weeks of low carb. We KNOW that weight loss slows over time (especially if calories are controlled, as they were in this study). It has to do with the order of treatments.
Next time someone says VLC/keto is harmful or at least not helpful for fat loss because of a new rodent study, they’ll probably be wrong.
BOOKMARK THIS ONE GUYS.
Rodent studies on ketogenic diets or exogenous ketones are valuable and interesting in a variety of #contexts, although I’d argue that regulation of fat mass isn’t really one of ’em.
For starters, rodents aren’t particularly ketogenic – it’s rare to see ketones >1 after an overnight fast even in long-term ketoadapted mice. Also, many rodents gain weight until they die, whereas humans plateau and stay relatively weight-stable for their entire lives (at least historically, and I’m not talking about yo-yo dieting).
Skeletal muscle, on the other hand, seems more similarly regulated: keto isn’t muscle-sparing in either specie… most people, perhaps unwittingly, increase protein intake on keto, and THIS spares muscle (N.B. this is simply to spare muscle, whereas in non-keto dieters, it’s not uncommon to see increased muscle in the #context of high protein). That’s because carbs are more anabolic than fat. QED.
There’s just a fundamental difference in the way fat mass and appetite is regulated between the species. There are many similarities, which is why these studies are still valuable, but fat mass isn’t one of ‘em.
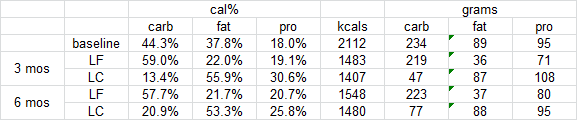
Weight loss on low-fat vs. low-carbohydrate diets by insulin resistance status among overweight and obese adults: a randomized pilot trial (Gardner et al., 2015)
Low carb diet: participants went from 230 grams/d to less than 50 for the first 3 months, then creeped up to ~80 over the next 3 months.
Will the critics say “the carbz weren’t low enough!”? REALLY?
Brief background reading: amylin (according to Wikipedia)
In a study by Hollander on type II diabetics, the synthetic amylin analog pramlintide was tested (Hollander et al., 2003). In this year-long RCT, over 600 patients were treated with placebo or up to 120 ug pramlintide BID (twice per day). On average, these subjects were obese (BMI 34), diabetic for ~12 years, and had an HbA1c of 9.1%. After one year, HbA1c declined 0.62% and they lost about 1.4 kg… not very impressive.
But it’s not all bad news; after viewing those relatively negative results (3 lb weight loss over the course of 1 year), another group of researchers led by Louis Aronne and Christian Weyer believed amylin had yet to be tested proper. So they designed a better study; it was shorter, used higher doses of pramlintide, and they enrolled obese yet non-diabetic patients (Aronne et al., 2007). They opted for higher doses of pramlintide (240 ug TID [three times per day]) because in dose-escalation studies, the incidence and severity of adverse drug reactions was consistently low at all doses tested.
They chose to study obese-er subjects (BMI 38, compared to 34 in the Hollander study) because obese subjects lose fat more readily than lean people, so if the study is designed to measure fat loss, then it is better to select a population of subjects where more fat loss is predicted. They selected non-diabetic subjects for a similar reason; diabetics must regularly inject insulin which promotes the accumulation of fat mass — this could counteract any fat reducing effects of pramlintide.
In other words, it was a more powerful and better designed study.
After 16 weeks, pramlintide-treated subjects lost an average of 3.6 kg (~8 lbs), or about half a pound per week. 30% of patients lost over 15 pounds (1 lb/wk)! Importantly, the weight loss didn’t appear to have reached a plateau by week 16, so it would have most likely continued along a similar trajectory had the study been longer. There were no side effects, and a battery of psychological evaluations showed that the patients receiving pramlintide felt it was easier to control their appetite and BW, they didn’t mind the daily injections, and overall well-being increased. At the very least, these evaluations meant the subjects weren’t losing weight because of nausea or malaise. In fact, it was quite the opposite.
Does it matter where fatty acids are oxidized, liver or skeletal muscle? Of course, they’re oxidized in both tissues (quantitatively much more in the latter), but relative increases in one or the other show interesting effects on appetite and the regulation of fat mass [in rodents].
Warning: a lot of speculation in this post.
A LOT.
It’s known that LC diets induce a spontaneous decline in appetite in obese insulin resistant patients. Precisely HOW this happens isn’t exactly known: the Taubes model? improved leptin signaling? probably a little bit of both, other mechanisms, and possibly this one:
If you’re healthy, no major complaints, then you probably won’t benefit from tweaking your ‘biome. Ymmv. But if you’re gonna do it anyway, here are some tips (mostly my opinions).
I came across a recent study on a mouse model of Angelman Syndrome (an epigenetic disorder), and wasn’t surprised to learn there’s a strong circadian component to it. Epigenetics are one of the main ways circadian rhythms are programmed.
In this case, the circadian connection is more direct.
Angelman Syndrome (AS): you inherit 2 pairs of each gene, one from Mom and one from Dad. In some cases, one of the copies is silenced via epigenetics and you’re basically just hoping the other one is in good shape. In the genetically relevant region in AS, the paternal copy is silenced and the maternal copy does all the heavy lifting, but in AS, the maternal copy is mutated or absent, so none of the genes in this region are expressed.
Interestingly, scientists found that one of the genes, Ube3a (an ubiquitin ligase), is involved in regulating Bmal1, a core circadian gene (Shi et al., 2015) . And mice with a silenced paternal Ube3a and mutant maternal Ube3a exhibit many of the same circadian symptoms of children with AS. They don’t mimic all of the symptoms as there are many other genes in this region. But both show circadian abnormalities.
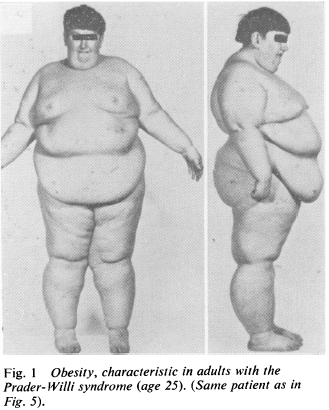
Prader-Willi Syndrome (PWS) is the epigenetic opposite: same region of DNA, but silenced maternal copy and mutant or absent paternal copy. This disorder is characterized by massive obesity and low muscle mass (among other things).
While reading about this disorder, I was taken aback with how the obesity was explained.
“Insatiable appetite” (Laurance et al., 1981), although from what I can gather, these children would develop massive obesity even if they were fed cardboard. Some studies even showed no change in food intake and/or energy expenditure (eg, Schoeller et al., 1988), which led some researchers to publish entire papers about how these children must be lying and/or stealing food (eg, Page et al., 1983) .
Further, other researchers even explained their obesity was due to an inability to vomit (Butler et al., 2007).
THEY’RE OBESE BECAUSE THEY’RE NOT BULEMIC.
AYFKM?
When these kids gain weight, it’s nearly all fat mass; when they lose weight, it’s nearly all muscle [shoulda been a BIG hint]… this even led some researchers (who detected no change in fat mass after significant weight loss) to conclude that their techniques to assess body composition must not be valid in this population because: surely, they must’ve lost some fat mass like normal people do.
THEY FAILED TO CONSIDER THIS IS AN EXTREME CIRCADIAN MISMATCH DISORDER IN NUTRIENT PARTITIONING
It was actually painful to read: these kids are being accused of stealing food and not vomiting because that’s the only way to explain it.
NO IT’S NOT, SCIENCE.
They can be forced into losing fat while maintaining some muscle with an extreme protein-sparing modified fast (eg, Bistrian et al., 1977)…
A few research groups have considered the possibility it’s a hormonal disorder, and some fairly long-term studies with GH replacement have shown promising results (eg, Carrel et al., 1999).
Prader-Willi Food Pyramid. Wait, wut? O_o
Some have even speculated involvement of leptin (eg, Cento et al., 1999), although this hasn’t been followed-up on.
Disclaimer: I don’t know the cure or best treatment modality for Prader-Willi, although given the strong circadian component in its sister condition, Angelman’s Syndrome, I strongly believe this avenue should be explored (in combination with the seemingly necessary hormonal corrections, which have been the only successful interventions yet). “Diet” doesn’t work; these kids aren’t obese because they’re stealing food or failing to vomit. Interventions strictly targeting CICO have massively failed this population.
Side note: in the Angelman Syndrome mouse model, *unsilencing* the paternal copy worked… maybe the same could work in PWS (and/or other forms of obesity)…?
Evidence supporting potential circadian-related treatment modalities for PWS:
A Prader-Willi locus IncRNA cloud modulates diurnal genes and energy expenditure (Powell et al., 2013)
Magel2, a Prader-Willi syndrome candidate gene, modulates the activities of circadian rhythm proteins in cultured cells (Devos et al., 2011)
Circadian fluctuation of plasma melatonin in Prader-Willi’s syndrome and obesity (Willig et al., 1986)
And the connection with LIGHT:
Artificial light at night: melatonin as a mediator between the environment and the epigenome (Haim and Zubidat, 2015)
Circadian behavior is light re-programmed by plastic DNA methylation (Azzi et al., 2014)
PWS is much worse than just nutrient partitioning (seriously, just spend a few minutes on any Prader-Willi support forum or this; maybe it is an appetite disorder, but given the data on weight gain [mostly fat mass] and weight loss [mostly muscle mass], it seems far more likely a circadian disorder of nutrient partitioning),
but that component jumped out at me; more specifically, despite the only positive results coming from non-dietary interventions, researchers were still all “#CICO.”
“Lean meat, sugar-free Jello, and skim milk”
FFS
Circadian biology, hormone replacement [where appropriate], and figure out if any specific diets help. PMSF/CR doesn’t work unless “refrigerators and cabinet pantries are locked shut.”
Maybe this applies to other forms of obesity, too.
Maybe.
Check out my Patreon campaign! Join the community of over 300 members for up-to-date information about a variety of topics in the health & optimizing wellness space. At 5 bucks a month, can’t beat it!
Affiliate links: KetoLogic for keto-friendly shakes, creamers, snacks, etc. And get 15% off your ketone measuring supplies HERE.
Still looking for a pair of hot blue blockers? TrueDarkis offering 10% off HERE and Spectra479is offering 15% off HERE. If you have no idea what I’m talking about, read this then this.
If you want the benefits of ‘shrooms but don’t like eating them, Real Mushrooms makes great extracts. 10% off with coupon code LAGAKOS. I recommend Lion’s Mane for the brain and Reishi for everything else.