
Dr. Johnson’s recent paper is nothing short of a monster. They did a TON of experiments. Here, I only want to focus on one aspect.
The insulin-obesity hypothesis in a nutshell (very oversimplified): more insulin = more fat mass and vice versa.
I know I know, it was mice fed standard rodent chow, but also included models relevant to human biology like reduced insulin and caloric restriction, which may reflect certain aspects of ketogenic diets and intermittent fasting… and some of the results actually do reflect what happens to humans. Some.
divide and conquer
They use two lines of mice: one with the insulin-2 gene deleted (Ins1+/+; Ins2-/-); and another that also had one copy of the insulin-1 gene deleted (Ins1+/-; Ins2-/-). The latter model has half the copies of insulin and for simplicity, I’ll refer to these as knockouts (KOs) and the others as wild-type (WT). Further, they also studied caloric restriction (CR) in both models.
Ergo, four groups: ad lib WT, ad lib KO, CR WT, and CR KO.
Interestingly, ad lib KOs had the lowest insulin. We may have expected CR KOs to have the lowest, but they don’t. And in agreement with the insulin-obesity theory, the CR KOs with more insulin also have more fat mass. Say what? The CR KO mice ate less (by design) but had more fat mass (supports insulin-obesity theory) (remember the fattier mice?).
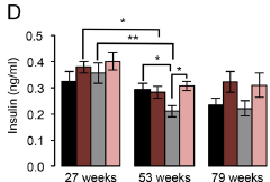

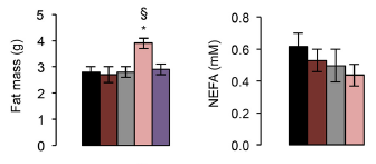
However, ad lib KOs had lower insulin than ad lib WT, but the same amount of fat mass. This either refutes the insulin-obesity hypothesis or, going out on a limb here, may suggest their adipose is more sensitive to insulin, so less insulin is required to induce the same amount of fat storage.
The authors hypothesized elevated GIP levels in ad lib KOs (relative to ad lib WT) may be responsible for this adipose tissue insulin sensitization. However, WT mice have the same level of insulin and fat mass regardless of ad lib or CR, despite CR having WAY more GIP. If GIP was responsible for adipose tissue insulin sensitization, then the same amount of insulin should’ve induced more fat growth, right?
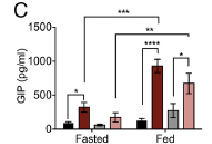
GIP in a nutshell: secreted from the upper small intestine in response to nutrients, insulinogenic, and fattening to the adipocyte.
In general, if leptin doesn’t at least roughly mirror fat mass, something’s up.
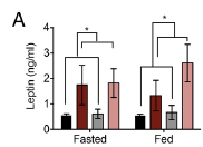
Something’s up.
In this study, CR KOs have the highest leptin and most fat mass, but CR WTs have more leptin than ad lib WTs (despite similar fat mass)… this may reflect an influence of the semi-starved state, so I’ll take it… #context and all that jazz.
Check out Jane Plain’s take on the GIP story here. Highly recommended. Infinitely quotable and ties in a lot of different #contexts including rebound weight-gain, intermittent fasting, and potential mechanisms mediating the effects.
It’s great, but the reasoning only seems to apply to ad lib WT and KOs. It doesn’t apply to CR and ad lib WTs, who have relatively the same amount of insulin as CR KOs. I mean, the difference in GIP between ad lib and CR WT is really big. CR WTs have the highest GIP of all and more insulin than ad lib KOs, but absolutely the same level of fat mass.
That’s TWO factors that should be promoting adipose accumulation, but aren’t.
So, my question is: what’s the #context mediating this disparity?
Also, highly recommend reading the paper — even the discussion section! Especially if you want to learn a lot about CR, GIP, energy balance, etc., in a short time.
For example, caloric restriction in the context of reduced insulin gene dosage… think: intermittent fasting and LCHF? or simply the spontaneous reduction in appetite observed by many on a ketogenic diet (yes, this is a form of CR). According to the Johnson-Plain Hypothesis™, there may be some people strongly predisposed to weight re-gain in these contexts.
Add in the findings of Gardner, Pittas, Cornier, and Ebbeling and we may be able to begin to hone in on who these people might be.