
Remember the “jet lag-resistant” mice? Guess what: screw with circadian biology and metabolism pays the price.
In brief, vasopressin was classically thought of as an anti-hypotensive hormone. The vasopressin analog Desmopressin is used to treat bed-wetting. But vasopressin biology is much more interesting than that: mice lacking both vasopressin receptors require very little time adapting to large circadian phase changes. And as with many fundamental concepts in chronobiology, this is intimately linked with metabolism.
People with certain polymorphisms of the vasopressin receptor, V1A, exhibit elevated blood glucose levels and are at greater risk for diabetes (Enhorning et al., 2009):
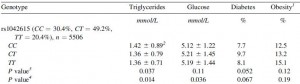
This risk is strongest in men in the highest quartile of fat intake, and is statistically more significant after adjusting for age and physical activity:
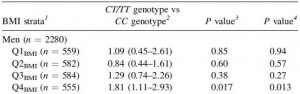
This study wasn’t designed to be a very powerful indicator of diet-disease relationships, but a little speculation: some think higher fat [and lower carb] intake should be protective against diabetes… which may be true, for people who can tell time. Alter one nucleotide in the vasopressin 1A receptor gene and the game changes.
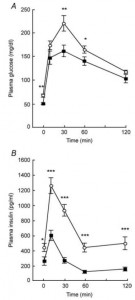
Removal of the vasopressin 1A receptor in mice mirrors the human polymorphism. Hyperglycemia and impaired glucose tolerance (Aoyagi et al., 2007)

Their livers crank out glucose in droves:

Aaaand when given an obesogenic diet, they eat just as much but gain significantly more weight:
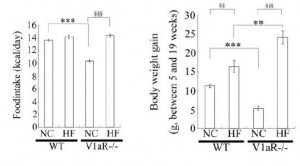
Odd, considering they’re fat-burning machines (Hiroyama et al., 2007), but similar to the human condition: mess with chronobiology and the game changes.
Remove V1B receptor, on the other hand, and the mice are “insulin hypersensitive” (Fujiwara et al., 2007):
…which may or may not be due to increased adiposity (Hiroyama et al., 2009):
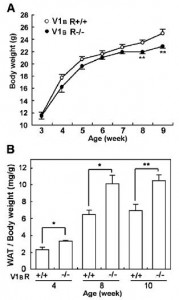
Remove both receptors, and they don’t simply cancel each other out (Nakamura et al., 2009): these mice eat less, weigh more:
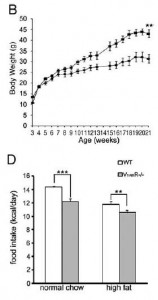
Removal of V1B increases adiposity which preserves insulin sensitivity; this doesn’t happen in the context of V1A deficiency… the double knockouts exhibit impaired glucose tolerance despite increased insulin = robust insulin resistance:
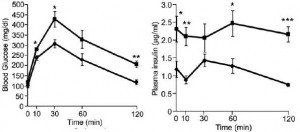
Summary
V1A KO: eat same, weigh more. Insulin resistant.
V1A polymorphism: increased risk of diabetes.
V1B KO: increased adiposity. “Insulin hypersensitive.”
V1AB KO: eat less, weigh more. Insulin resistant, yet seemingly impervious to circadian phase changes ->
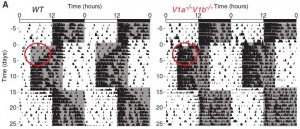
In the figure below, the black marks reflect activity. Mice are normally more active in the dark phase. Shift the dark phase 8 hours earlier (“jet lag”), and normal mice take about a week or so to adjust their circadian activity behavior (see red circle on left). However, V1AB KO’s adjust practically instantaneously (see red circle on right):
However, in contrast to the diverging effects on insulin sensitivity, both single knockouts appear slightly better at reentraining:
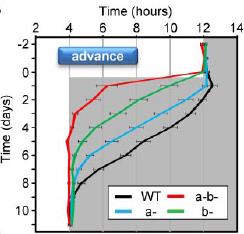
V1A KO: insulin resistant, pretty good at reentraining
V1B KO: insulin sensitive, a little better at reentraining
V1AB KO: insulin resistant, very good at reentraining
In each of these models, superior response to jet lag correlates with poor nutrient partitioning. Cause-effect? Maybe it’s supposed to take a while to adjust to jet lag, and we’re doing more harm than good by expediting adaptation to circadian phase changes via blue blockers, bright light therapy, etc… or we’re just not meant to fly long distances.
Alternatively, maybe it’s not “superior response to jet lag,” but rather these mice just have a “loose” clock: they can easily adapt to circadian phase changes because they’re not strongly tied into their current circadian phase, but by the same mechanism, nutrient partitioning is impaired. Or nutrient partitioning pays the price.
I just flew in from Albuquerque, and boy are my arms tired…and fat. http://t.co/870apbQeHw
— Gerard Pinzone (@GerardPinzone) August 12, 2014